
Help Home > Clustering > Phylogenetic Cluster Reconstruction
![]()
- Clustering Front Page
- Cluster Details Page
- Phylogenetic Cluster Reconstruction
Phylogenetic Cluster Reconstruction
For selected gene clusters, the JGI has attempted to organize cluster members into a phylogenetic tree. The default protocol for generating this tree is as follows: Protein sequences from a given gene cluster are aligned using MAFFT, with final alignments generated using Gblocks to identify high-reliability positions. When sufficient high-reliabililty positions are found, RAxML is used to generate gene trees based on those positions, and Notung is used to reconcile the gene tree with the species tree, which can be found in the Downloads section. Where the tree generation algorithm deviates from this protocol, it will be described in the Statistics panel (see below).
The Cluster Tree page is organized in three sections:
- 1. The Phylogenetically reconstructed gene tree (if available).
- 2. Statistics and information about the reconstruction.
- 3. Downloads of text versions of the tree and the alignments used in its construction.
1. The Gene Tree
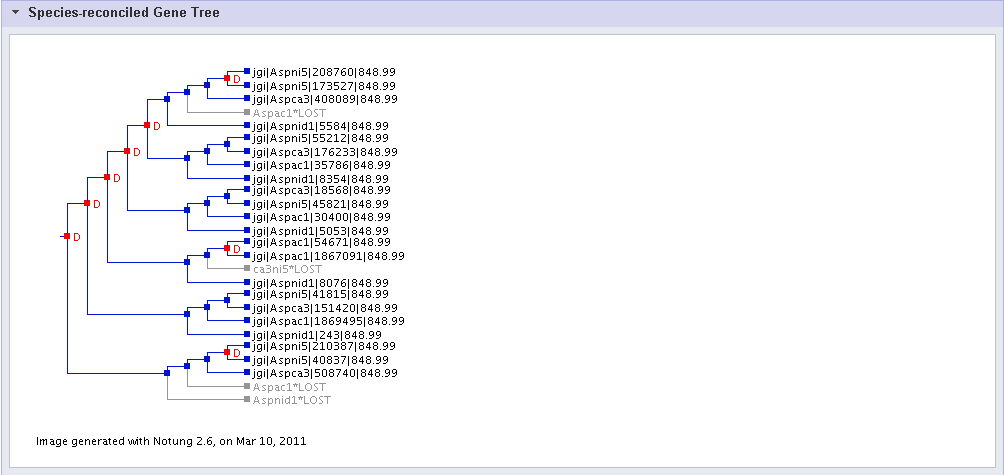
The phylogenetic tree reconstruction attempts to place each gene member of the cluster within the evolutionary tree defined by the various species that comprise the cluster.
A red D at a branch point indicates a predicted gene duplication within the cluster.
The symbol "*LOST" in grey indictes a predicted loss in a gene family. Note that a loss can be predicted for an individual gene within a single species (a "leaf" node) or for a non-leaf node within the species tree. In the latter case, descendants of the lost node will not be shown (the lost descendant nodes are shown in the complete species tree for the cluster, which can be found in the Downloads section (see below).
2. Reconstruction Statistics

- The number of species and genes represented in the cluster.
- The number of amino acid positions conserved among genes in the cluster that were used for tree generation in the calculated alignment.
- The number of gene duplication and loss events predicted by the tree reconstruction.
- A description of the algorithm used to generate the tree.
3. Downloads
- Cluster Fasta sequences. Amino acid sequences of the cluster members.
- Raw Alignment (fasta format). Inital MAFFT alignment of cluster sequences
- Final Alignment (fasta format). Final alignment generated by Gblocks.
- Unreconciled gene tree (nhx format). The gene tree defined by the multiple alignment alone (without species information).
- Species tree image. a png image of the tree.
- Species tree data file. The data file defining the tree in nhx format.
- Final/species-reconciled gene tree. The final tree in nhx format.