|
Help Home > The Genome Browser > What is a Scaffold? What is a Scaffold? |
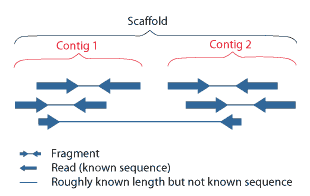
| A scaffold is a portion of the genome sequence reconstructed from end-sequenced whole-genome shotgun clones. Scaffolds are composed of contigs and gaps. A contig is a contiguous length of genomic sequence in which the order of bases is known to a high confidence level. Gaps occur where reads from the two sequenced ends of at least one fragment overlap with other reads in two different contigs (as long as the arrangement is otherwise consistent with the contigs being adjacent). Since the lengths of the fragments are roughly known, the number of bases between contigs can be estimated.
The goal of whole-genome shotgun assembly is to represent each genomic sequence in one scaffold; however, this is not always possible. One chromosome may be represented by many scaffolds (e.g., Chlamydomonas reinhardtii) or just a single scaffold (e.g., Human chromosome 19), depending on how completely the genome can be reconstructed, or assembled, from the available reads. The relative locations of scaffolds in the genome are unknown. Scaffolds are normally numbered approximately from largest to smallest. Some scaffolds may ultimately be filtered out of the assembly, resulting in skipped scaffold numbers. In some cases, scaffolds can overlap. For example, in polymorphic genomes, regions with a high density of allelic differences between haplotypes may be split into separate sets of scaffolds, each representing one allele. Thus, a sequence that exists in only one location in the genome may appear on more than one scaffold. Gaps are shown in the Genome Viewer as red lines or rectangles in the scaffold track (viewed in "full" mode). Contigs are shown in black. In FASTA sequences, gaps are represented by a series of Ns. |